LoadBatch: Difference between revisions
Created page with 'LoadExperiment enables you to load a set of experimental quantitative, polymorphism, or alignment data for a genome in CoGe. Several different file formats are supported. The dat...' |
No edit summary |
||
| Line 1: | Line 1: | ||
<div style="color:red"> | |||
UNDER CONSTRUCTION | |||
</div> | |||
[[File: | LoadBatch provides the ability to conveinently load a set of genomes or experiments in a single operation. To load a set of genomes using [[LoadGenome]] would require running the tool for each genome individually. | ||
[[File:LoadBatch.png|thumb|400px]] | |||
== Inputs == | == Inputs == | ||
| Line 7: | Line 11: | ||
=== Metadata === | === Metadata === | ||
=== Data File === | === Data File === | ||
Revision as of 17:09, 16 September 2014
UNDER CONSTRUCTION
LoadBatch provides the ability to conveinently load a set of genomes or experiments in a single operation. To load a set of genomes using LoadGenome would require running the tool for each genome individually.

Inputs
Metadata
Data File
You can select and retrieve data file located at:
- The iPlant Data Store
- An FTP server
- Your computer (Upload)
Data Formats and Track Types
LoadExperiment supports several data file formats depending on the data type:
- Quantitative data
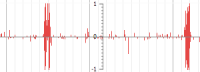
Quantitative track - Comma-separated (CSV) file format
- Tab-separated (TSV) file format
- BED file format
- Marker data

Marker track - GFF/GTF file format
- Polymorphism (SNP) data

SNP track - Variant Call Format (VCF) file format
- Alignment data
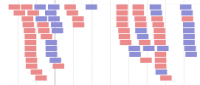
Alignment track - BAM file format




Each of these file formats are described below in their own section. The file type can be auto-detected by LoadExperiment if the file name ends with the expected extension (.csv, .tsv, .bed, .gff, .gtf, .vcf, .bam). Files can be compressed (.zip, .gz) and still have their type auto-detected (e.g., mydata.bed.gz). For non-standard file name extensions, you can select the file type from a list.
CSV File Format
This is a comma-delimited file that contains the following columns
- Chromosome (string)
- Start position (integer)
- Stop position (integer)
- Chromosome Strand (1 or -1)
- Measurement Value must be between [1-0] (real number; inclusive)
- Second Value (OPTIONAL): can store a second value such as an expect value (real number)
#CHR,START,STOP,STRAND,VALUE1(0-1),VALUE2(ANY-ANY) Chr1,11486,12316,1,0.181430277220112,7.3980806218146 Chr1,27309,28272,1,0.944373742485446,5.08225285439412 Chr1,32484,32978,1,0.328500324191726,1.97719838086201 Chr1,41942,42508,-1,0.825027233105203,6.56057592312617 Chr1,56394,57527,-1,0.183234367788511,0.795527328556531 Chr1,67705,68809,-1,0.956523086778851,5.20992343466606 Chr1,71144,72409,1,0.42955128220331,1.80604269639474 Chr1,81671,82833,1,0.626003507696723,2.77834108023821 Chr1,86467,87623,-1,0.0878653961575928,7.42843749315945
TSV File Format
Same as CSV format but with tab delimiters instead of commas.
BED File Format
Standard BED format as defined here: http://genome.ucsc.edu/FAQ/FAQformat.html#format1
Only the first six columns are used, with the "name" field ignored.
GFF File Format
Standard GFF3 format as defined here: http://gmod.org/wiki/GFF3
Only the seqid, start, end, score, strand, and attribute columns are used (column numbers 1, 4, 5, 6, 7, 9 respectively).
VCF File Format
Standard VCF 4.1 format as defined here: http://www.1000genomes.org/wiki/Analysis/Variant%20Call%20Format/vcf-variant-call-format-version-41
BAM File Format
Standard BAM format.
FASTQ Data
EPIC-CoGe now supports fastq data generated by RNASeq. When loaded, EPIC-CoGe will run and the Expression Analysis Pipeline developed by James Schnable for his qTeller project.
Bulk Loading
Please contact the CoGe Team if you have large numbers of experiments you wish to load and we can help you with the bulk loading.