LoadExperiment
LoadExperiment enables you to load experimental quantitative, polymorphism, or alignment data for a genome in CoGe. Many common file formats are supported. The data can then be viewed alongside genome sequence/annotation in GenomeView. The LoadExperiment "wizard" guides you through each step:
- Describe your experiment
- Add data
- Select Options
- Review and Submit
Metadata: describe your experiment
The first step in loading an experiment into CoGe is to describe it with metadata. CoGe requires a minimum set of metadata fields:

- Name: Name of experiment
- Description: Description of experiment
- Version: Version of experiment. Can be a combination of numbers and letters. Exclude the leading "v" as CoGe will add that for you. Set to "1" if you aren't sure what the value should be.
- Source: Where is the data from? This could be you, your lab, your university, a sequencing center, your collaborator.
- Restricted: Specify whether the experiment public or restricted to you and your collaborators
- Genome: Select the appropriate genome from CoGe to associate the data with
- Note: Additional metadata about the experiment can be added as well.
- Example from an experiment loaded into EPIC-CoGe: http://genomevolution.org/CoGe/ExperimentView.pl?eid=193
- Information on providing a metadata file for bulk import: Experiment Metadata
Add primary data

You can select and retrieve data file located at:
- The iPlant Data Store
- CoGe has automatic access to the 'coge_data' directory in your iPlant Data Store. You can place your data to load into that directory, or manually share the data with the "coge" user.
- An FTP or HTTP URL
- Your computer (Upload)
- Not recommended for large files!
Note: Typically only a single data file can be selected. The exception is FASTQ files for which multiple files can be selected.
Data Formats and Track Types
LoadExperiment supports several data file formats depending on the data type:
- Quantitative data
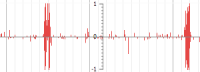
Quantitative track - Comma-separated (CSV) file format
- Tab-separated (TSV) file format
- BED & BedGraph file formats
- WIG & BigWig file format
- Marker data

Marker track - GFF/GTF file format
- Polymorphism (SNP) data

SNP track - Variant Call Format (VCF/GVCF) file format
- Alignment data
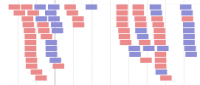
Alignment track - BAM file format
- FASTQ file format




Each of these file formats are described below in their own separate section. The file type can be auto-detected by LoadExperiment if the file name ends with the expected extension (.csv, .tsv, .bed, .gff, .gtf, .vcf, .bam). Files can be compressed (.zip, .gz) and still have their type auto-detected (e.g., mydata.bed.gz). For non-standard file name extensions, you can force a specific type by selecting it from the list of file types.
CSV File Format
This is a comma-delimited file that contains the following columns
- Chromosome (string)
- Start position (integer)
- Stop position (integer)
- Chromosome Strand (1 or -1)
- Measurement Value (real number)
- Second Value (OPTIONAL): can store a second value such as an expect value (real number)
#CHR,START,STOP,STRAND,VALUE1,VALUE2 1,11486,12316,1,0.181430277220112,7.3980806218146 1,27309,28272,1,0.944373742485446,5.08225285439412 1,32484,32978,1,0.328500324191726,1.97719838086201 1,41942,42508,-1,0.825027233105203,6.56057592312617 1,56394,57527,-1,0.183234367788511,0.795527328556531 1,67705,68809,-1,0.956523086778851,5.20992343466606 1,71144,72409,1,0.42955128220331,1.80604269639474 1,81671,82833,1,0.626003507696723,2.77834108023821 1,86467,87623,-1,0.0878653961575928,7.42843749315945
TSV File Format
Same as CSV format but with tab delimiters instead of commas.
BED & BedGraph File Formats
- Standard BED format as defined here: http://genome.ucsc.edu/FAQ/FAQformat.html#format1
- Only the first six columns are used, with the "name" field ignored.
- BedGraph format as defined here: http://genome.ucsc.edu/goldenPath/help/bedgraph.html
Note that the 0-based coordinates of the BED format will be translated into 1-based within CoGe.
WIG & BigWig File Format
- Standard WIG format as defined here: http://genome.ucsc.edu/goldenpath/help/wiggle.html
- Only the "variableStep" type is supported. The "fixedStep" type is not supported and will cause the load to fail with an error.
- Start and end coordinates are expected to be 1-based.
- Standard BigWig format as defined here: https://genome.ucsc.edu/goldenpath/help/bigWig.html
- UCSC's bigWigToWig program is used to convert the BigWig file to a WIG file for loading.
GFF File Format
Standard GFF3 format as defined here: http://gmod.org/wiki/GFF3
Only the seqid, start, end, score, strand, and attribute columns are used (column numbers 1, 4, 5, 6, 7, 9 respectively).
VCF File Format
Standard VCF 4.1 format as defined here: http://www.1000genomes.org/wiki/Analysis/Variant%20Call%20Format/vcf-variant-call-format-version-41
In short, it is a tab-separated file that is separated into two sections:
- Metadata information lines at the top of the file
- The sequencing data set containing all of the comparisons of the individual(s) to a reference genome with the following column headings:
- Chromosome - name of the scaffold, pseudomolecule, or chromosome the SNP call is located on
- Position - where the SNP is called on the chromosome in bp
- ID - a semicolon separated list of unique identifiers for the SNP. If there is no identifier a period (".") should be recorded. No white space or semicolons.
- Reference - the base that exists on the reference genome (A,G,C,T,N). Multiple nucleotides are shown for indels or complex substitutions.
- Alt - comma separated list of nucleotides that are different in an individual from the reference (A,G,C,T,N,*). The asterisk denotes a deletion.
- Quality - Phred quality score for the alternate nucleotide call.
- Filter - whether the ref-alt pair passed any filters set in analysis. The word 'pass' will be present if the section passes all filters. A blank represented by a period ('.') will show if no filters were used
- Info -
- Format -
- Individuals - can be any number of columns describing each individual in the population of interest
GVCF for Population Genetics Calculations
Chromosome Position ID Reference Alt Quality Filter INFO Format Indiv1 Indiv2 scaffold_1 114 . A . 52.8 . . GT:DP 0/0:23 0/0:19 scaffold_1 116 . C T 151.24 . . GT:AD:DP:GQ:PL 0/0:23,0:23:54:0,54,392 0/0:17,0:19:24:0,24,231 scaffold_1 117 . C A 704.91 . . GT:AD:DP:GQ:PL 0/0:25,0:25:57:0,57,413 0/0:18,2:20:8:0,8,193 scaffold_1 118 . T . 52.83 . . GT:DP 0/0:25 0/0:20 scaffold_1 119 . A . 14.95 LowQual . GT:DP 0/0:26 0/0:20 scaffold_1 120 . T G 250.44 . . GT:AD:DP:GQ:PL 0/1:16,3:19:8:8,0,72 0/1:29,10:40:77:77,0,318 scaffold_1 121 . A T 49.94 . . GT:AD:DP:GQ:PL 0/0:39,0:39:63:0,63,473 0/0:18,0:18:6:0,6,43 scaffold_1 122 . C . 15.89 LowQual . GT:DP 0/0:28 0/0:20 scaffold_1 123 . C . 49.89 . . GT:DP 0/0:29 0/0:20
BAM File Format
Standard BAM format.
FASTQ Data
RNAseq reads in FASTQ format.
- Single-ended: one or more single-ended FASTQ files can be specified.
- Paired-end: only two paired-end FASTQ files can be specified. The filename corresponding to the 5' reads should end with "R1" and the filename for the 3' reads should end with "R2".
- Example: Sample1_R1.fastq, Sample2_R2.fastq
Select Options for FASTQ data processing

This step displays options specific to the type of data that you selected.
The most complicated options are for FASTQ input files. CoGe provides multiple downstream analyses in addition to mapping reads to the reference sequence.
Expression Analysis Pipeline
The Expression Analysis Pipeline developed by James Schnable for his qTeller project.
SNP Identification Pipeline
Multiple methods of SNP identification are available to choose from:
- CoGe method: a naive SNP detection method that extracts sites from a SAMtools mpileup file that meet certain minimum requirements
- Platypus: popular SNP software from the Wellcome Trust Centre, http://www.well.ox.ac.uk/platypus
- SAMtools: a method using the SAMtools software, http://samtools.sourceforge.net/mpileup.shtml
- GATK: currently not yet available ...stay tuned! https://www.broadinstitute.org/gatk/guide/topic?name=tutorials#tutorials2803
Methylation Analysis Pipeline
Analyze whole genome bisulfite sequencing (WGBS) reads for methylation analysis using the Methylation Analysis Pipeline
The methylation pipeline was developed by Jeff Grover at the University of Arizona.
The metaplot analysis was developed by Josquin Daron and Keith Slotkin at Ohio State University.
ChIP-seq Analysis Pipeline
Analyze chromatin immunoprecipitation sequence using the ChIP-seq Analysis Pipeline.
Review and Submit
This final step simply displays of summary of the metadata, data, and options you've selected. To begin the load click the "Start Loading" button.
Troubleshooting
Make sure that these basic requirements are followed:
- The file format must be one of those listed above.
- The chromosome names used in the data files exactly match those for the genome. Mismatches will cause the load to fail.
- For text files, such as .CSV and .VCF, the newline (EOL) characters must be Unix-compliant (LF character, not CRLF) , see this article. This is sometimes a problem when loading text files created in Excel on Windows OS.
Bulk Loading
Please contact the CoGe Team if you have large numbers of experiments you wish to load and we can help you with the bulk loading.
Demo Data
These data are smaller than "real" datasets, but allow you to learn/test the system quickly (or use for demo's teaching)
- TODO